4. Genome assembly¶
4.1. Preface¶
In this section we will use our skill on the command-line interface to create a genome assembly from sequencing data.
Note
You will encounter some To-do sections at times. Write the solutions and answers into a text-file.
4.2. Overview¶
The part of the workflow we will work on in this section can be viewed in Fig. 4.1.
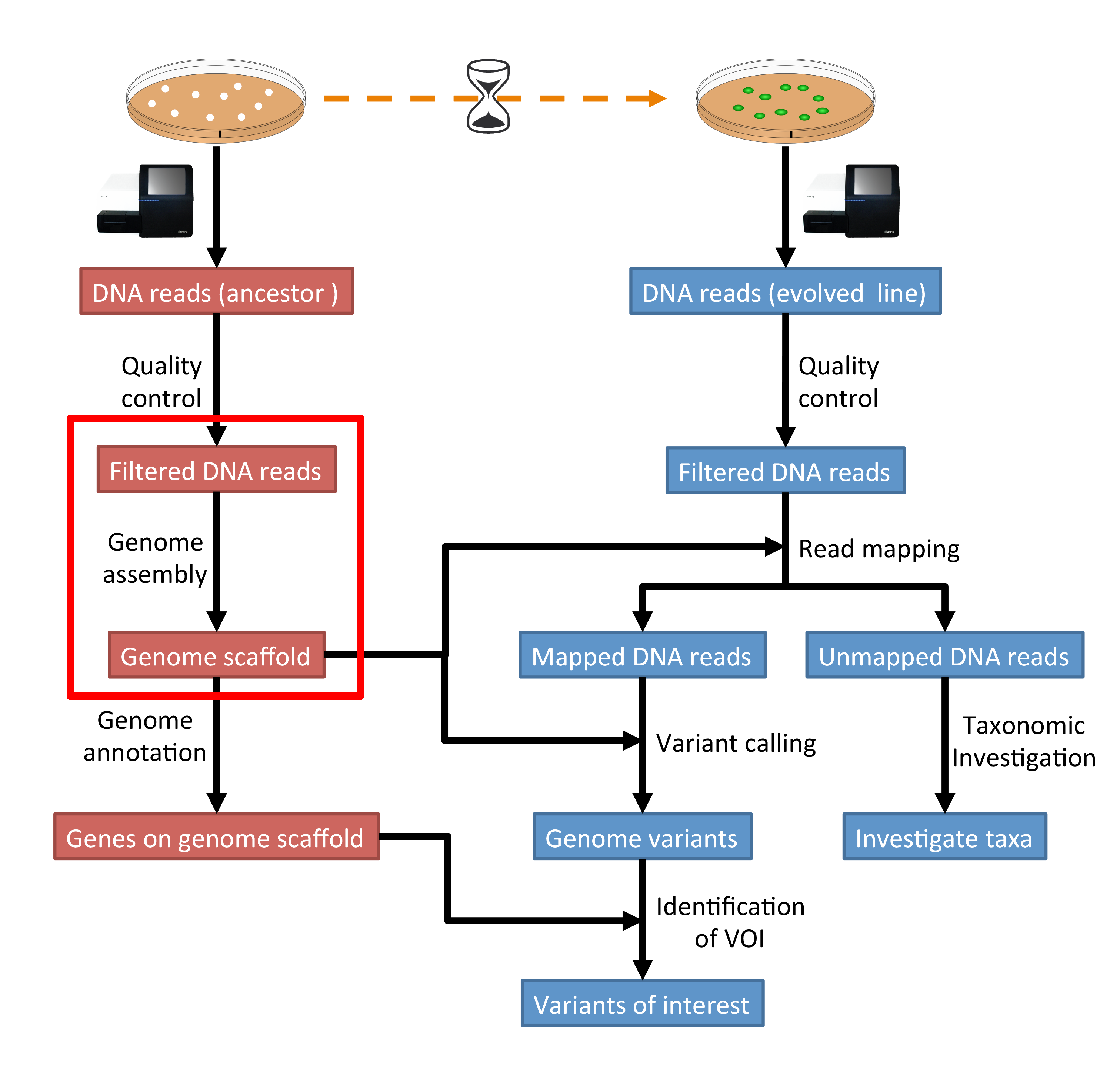
Fig. 4.1 The part of the workflow we will work on in this section marked in red.¶
4.3. Learning outcomes¶
After studying this tutorial you should be able to:
Compute and interpret a whole genome assembly.
Judge the quality of a genome assembly.
4.4. Before we start¶
Lets see how our directory structure looks so far:
cd ~/analysis
ls -1F
data/
trimmed/
trimmed-fastqc/
4.4.1. Subsampling reads¶
Due to the size of the data sets you may find that the assembly takes a lot of time to complete, especially on older hardware. To mitigate this problem we can randomly select a subset of sequences we are going to use at this stage of the tutorial. To do this we will install another program:
conda activate ngs
conda install seqtk
Now that seqtk has been installed, we are going to sample 10% of the original reads:
# change directory
cd ~/analysis
# create directory
mkdir sampled
# sub sample reads
seqtk sample -s11 trimmed/ancestor-R1.trimmed.fastq.gz 0.1 | gzip > sampled/ancestor-R1.trimmed.fastq.gz
seqtk sample -s11 trimmed/ancestor-R2.trimmed.fastq.gz 0.1 | gzip > sampled/ancestor-R2.trimmed.fastq.gz
In the commands below you need to change the input directory from trimmed/ to sampled/.
Note
The -s options needs to be the same value for file 1 and file 2 to samples the reads that belong to each other. It specified the seed value for the random number generator.
Note
It should be noted that by reducing the amount of reads that go into the assembly, we are loosing information that could otherwise be used to make the assembly. Thus, the assembly will be likely “much” worse than when using the complete dataset.
4.5. Creating a genome assembly¶
We want to create a genome assembly for our ancestor. We are going to use the quality trimmed forward and backward DNA sequences and use a program called SPAdes to build a genome assembly.
Todo
Discuss briefly why we are using the ancestral sequences to create a reference genome as opposed to the evolved line.
4.5.1. Installing the software¶
We are going to use a program called SPAdes fo assembling our genome. In a recent evaluation of assembly software, SPAdes was found to be a good choice for fungal genomes [ABBAS2014]. It is also simple to install and use.
conda activate ngs
conda install spades
4.5.2. SPAdes usage¶
# change to your analysis root folder
cd ~/analysis
# first create a output directory for the assemblies
mkdir assembly
# to get a help for spades and an overview of the parameter type:
spades.py -h
The two files we need to submit to SPAdes are two paired-end read files.
spades.py -o assembly/spades-default/ -1 trimmed/ancestor-R1.trimmed.fastq.gz -2 trimmed/ancestor-R2.trimmed.fastq.gz
Todo
Run SPAdes with default parameters on the ancestor
Read in the SPAdes manual about about assembling with 2x150bp reads
Run SPAdes a second time but use the options suggested at the SPAdes manual section 3.4 for assembling 2x150bp paired-end reads (are fungi multicellular?). Use a different output directory
assembly/spades-150for this run.
Hint
Should you not get it right, try the commands in Code: SPAdes assembly (trimmed data).
4.6. Assembly quality assessment¶
4.6.1. Assembly statistics¶
Quast (QUality ASsesment Tool) [GUREVICH2013], evaluates genome assemblies by computing various metrics, including:
N50: length for which the collection of all contigs of that length or longer covers at least 50% of assembly length
NG50: where length of the reference genome is being covered
NA50 and NGA50: where aligned blocks instead of contigs are taken
missassemblies: misassembled and unaligned contigs or contigs bases
genes and operons covered
It is easy with Quast to compare these measures among several assemblies. The program can be used on their website.
conda install quast
Run Quast with both assembly scaffolds.fasta files to compare the results.
Note
Should you be unable to run SPAdes on the data, you can manually download the assembly from Downloads. Unarchive and uncompress the files with tar -xvzf assembly.tar.gz.
quast -o assembly/quast assembly/spades-default/scaffolds.fasta assembly/spades-150/scaffolds.fasta
Todo
Compare the results of Quast with regards to the two different assemblies.
Which one do you prefer and why?
4.7. Compare the untrimmed data¶
Todo
To see if our trimming procedure has an influence on our assembly, run the same command you used on the trimmed data on the original untrimmed data.
Run Quast on the assembly and compare the statistics to the one derived for the trimmed data set. Write down your observations.
Hint
Should you not get it right, try the commands in Code: SPAdes assembly (original data).
4.8. Assemblathon¶
Todo
Now that you know the basics for assembling a genome and judging their quality, play with the SPAdes parameters and the trimmed data to create the best assembly possible. We will compare the assemblies to find out who created the best one.
Todo
Once you have your final assembly, rename your assembly directory int
spades_final, e.g.mv assembly/spades-default assembly/spades_final.Write down in your notes the command used to create your final assembly.
Write down in your notes the assembly statistics derived through Quast
4.9. Further reading¶
4.9.1. Background on Genome Assemblies¶
How to apply de Bruijn graphs to genome assembly. [COMPEAU2011]
Sequence assembly demystified. [NAGARAJAN2013]
4.9.2. Evaluation of Genome Assembly Software¶
GAGE: A critical evaluation of genome assemblies and assembly algorithms. [SALZBERG2012]
Assessment of de novo assemblers for draft genomes: a case study with fungal genomes. [ABBAS2014]
4.10. Web links¶
Lectures for this topic: Genome Assembly: An Introduction
Bandage (Bioinformatics Application for Navigating De novo Assembly Graphs Easily) is a program that visualizes a genome assembly as a graph [WICK2015].
References
- ABBAS2014(1,2)
Abbas MM, Malluhi QM, Balakrishnan P. Assessment of de novo assemblers for draft genomes: a case study with fungal genomes. BMC Genomics. 2014;15 Suppl 9:S10. doi: 10.1186/1471-2164-15-S9-S10. Epub 2014 Dec 8.
- COMPEAU2011
Compeau PE, Pevzner PA, Tesler G. How to apply de Bruijn graphs to genome assembly. Nat Biotechnol. 2011 Nov 8;29(11):987-91
- GUREVICH2013
Gurevich A, Saveliev V, Vyahhi N and Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 2013, 29(8), 1072-1075
- NAGARAJAN2013
Nagarajan N, Pop M. Sequence assembly demystified. Nat Rev Genet. 2013 Mar;14(3):157-67
- SALZBERG2012
Salzberg SL, Phillippy AM, Zimin A, Puiu D, Magoc T, Koren S, Treangen TJ, Schatz MC, Delcher AL, Roberts M, Marçais G, Pop M, Yorke JA. GAGE: A critical evaluation of genome assemblies and assembly algorithms. Genome Res. 2012 Mar;22(3):557-67
- WICK2015
Wick RR, Schultz MB, Zobel J and Holt KE. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 2015, 10.1093/bioinformatics/btv383